近日,数学与统计学院孙剑教授团队在人工智能辅助化学反应研究方面取得重要进展。研究团队提出基于距离几何空间的化学反应过渡态生成模型TS-DFM,相关成果以“Generative Flow Model on Distance Geometry for Predicting Transition States of Chemical Reactions”为题,在线发表于《自然·通讯》(Nature Communications),论文作者为西安交通大学数学与统计学院骆宇飞(博士生)、古祥(助理教授)、孙剑(教授)。
论文链接:https://www.nature.com/articles/s41467-026-74101-0
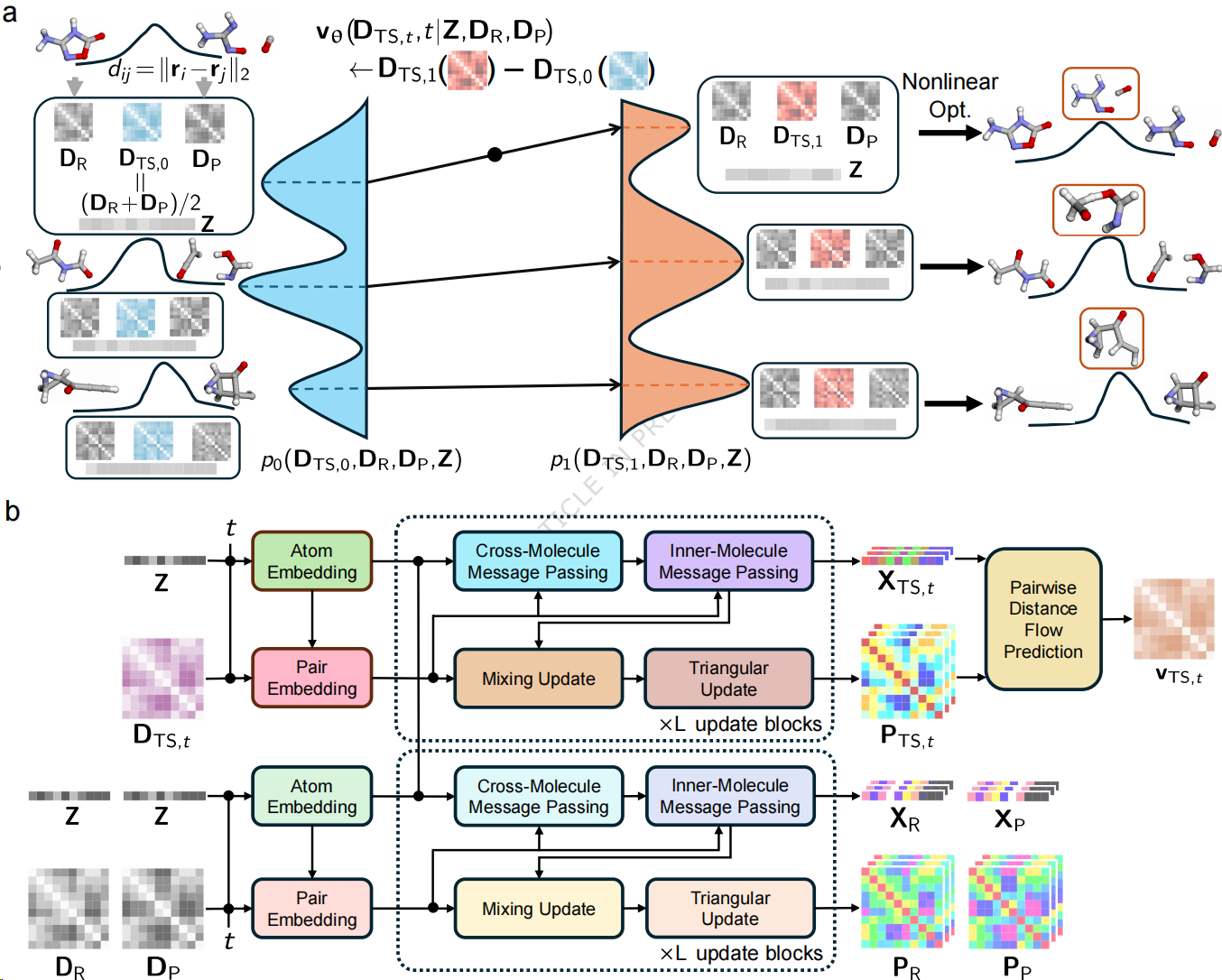
过渡态是连接化学反应物与产物的关键状态,其能量和结构直接影响化学反应速率与反应路径,是理解反应机理、设计催化剂、优化反应条件和开发新型合成路线的重要基础。然而,过渡态存在时间极短,难以通过实验直接捕捉。传统方法通常需要借助量子化学计算和迭代搜索获得过渡态结构,不仅计算成本较高,而且容易受到初始结构和收敛条件的影响。近年来,人工智能方法为过渡态预测提供了新的技术路径,但现有方法大多直接在三维坐标空间中生成分子结构,难以充分刻画化学反应过程中化学键断裂、形成和重组的内在规律。
针对上述问题,研究团队提出TS-DFM方法,将生成流模型与分子距离几何表示相结合,把过渡态预测转化为原子间距离变化的生成建模问题。与直接预测原子三维坐标不同,该方法重点学习反应过程中原子间距离以及化学键结构的演化,使模型的生成过程更加符合化学反应的物理规律,并减少原子碰撞等不合理中间结构的产生。TS-DFM采用基于最优传输的条件流匹配方法,以反应物和产物的距离信息作为模型输入,并通过双分支神经网络TSDVNet融合原子类型、反应物结构和产物结构等信息,逐步生成目标过渡态的距离矩阵。在获得距离信息后,模型通过非线性优化恢复过渡态的三维分子结构,从而将人工智能的生成能力与化学反应中的几何约束有机结合。
研究团队在Transition1x和RGD1等化学反应数据集上对TS-DFM进行了系统验证。实验结果显示,与现有代表性方法相比,TS-DFM在过渡态结构预测精度方面取得明显提升,在Transition1x数据集上的平均结构预测精度较最优基准方法提高约30%。模型预测得到的过渡态还能够作为传统CI-NEB过渡态搜索方法的高质量初始结构,有效减少后续计算所需的迭代次数,提高搜索算法的收敛速度和整体计算效率。除准确预测已有过渡态外,TS-DFM还表现出较好的泛化能力和反应路径探索能力。在训练数据中未出现的反应类型上,该方法的预测性能较现有方法提升至少16%;通过对反应物和产物结构进行适当扰动,模型还可以生成多个候选过渡态,并结合后续计算发现潜在的替代反应路径,为探索复杂化学反应机制提供了新的技术手段。
该研究为人工智能方法与化学反应物理规律的深度融合提供了新的思路,也为化学反应过渡态预测、反应路径搜索和反应机理分析提供了高效的智能化工具。未来,该方法有望进一步拓展至催化反应、材料化学和生物分子反应等复杂体系,为催化剂设计、反应网络构建和新型化学反应发现提供方法支持。


